


В Ильинскую больницу обратился мужчина, 66 лет, с жалобами на снижение уровня гемоглобина, слабость.
При сборе анамнеза стало известно, что четыре года назад (в 62 года) впервые стали беспокоить немотивированные подъемы температуры тела до 39,0, боли в суставах. Был неоднократно обследован амбулаторно и стационарно, но диагноз не удавалось верифицировать.
Три года назад появилась боль в ушных раковинах, и был поставлен диагноз полихондрит, далее был тромбоз правой бедренной вены, панникулит, пневмония с гидротораксом.
Год назад стала беспокоить анемия, что требовало регулярных трансфузий эритроцитарной массы, а в настоящее время – еще и тромбоцитопения.
При проведении молекулярно-генетического обследования выявлена соматическая мутация в гене UBA1, что позволило подтвердить сложный редкий диагноз – синдром VEXAS, который относится к аутовоспалительным заболеваниям.
Сейчас пациент проходит дообследование для дальнейшего назначения патогенетической терапии.
В Ильинской больнице впервые в стране организован центр диагностики и лечения аутовоспалительных заболеваний. В рамках Ильинской больницы можно реализовать полное обследование на редкие аутовоспалительные заболевания, включая соматические мутации. Ведь правильно поставленный диагноз – прямой путь к выздоровлению!
Синдром VEXAS
Синдром VEXAS впервые был описан совсем недавно, в 2020 году. David Beck с коллегами выявили соматическую мутацию в гене UBA1 в ходе ретроспективного исследования большой базы данных системы здравоохранения США.
VEXAS - аббревиатура, основанная на ключевых особенностях синдрома:
V (vacuoles) - вакуоли в миелоидных и эритроидных клетках костного мозга;
E1 - фермент, относящийся к убиквитин-активирующему ферменту, кодируемому UBA1 в Х-хромосоме;
A (autoinflammatory disease) - аутовоспаление;
S (somatic mutation) - соматическая мутация.
Синдром VEXAS встречается у мужчин старше 50 лет, так как мутация локализована в X-хромосоме и проявляется соматически, то есть возникает в течение жизни, а не наследуется.
Патогенез связан с дефектом в гене UBA1, который кодирует фермент Е1, участвующий в процессе убиквитинации, что приводит к дефициту фермента Е1 в миелоидных и эритроидных предшественниках клеток костного мозга и вызывает активацию аутовоспалительного процесса.
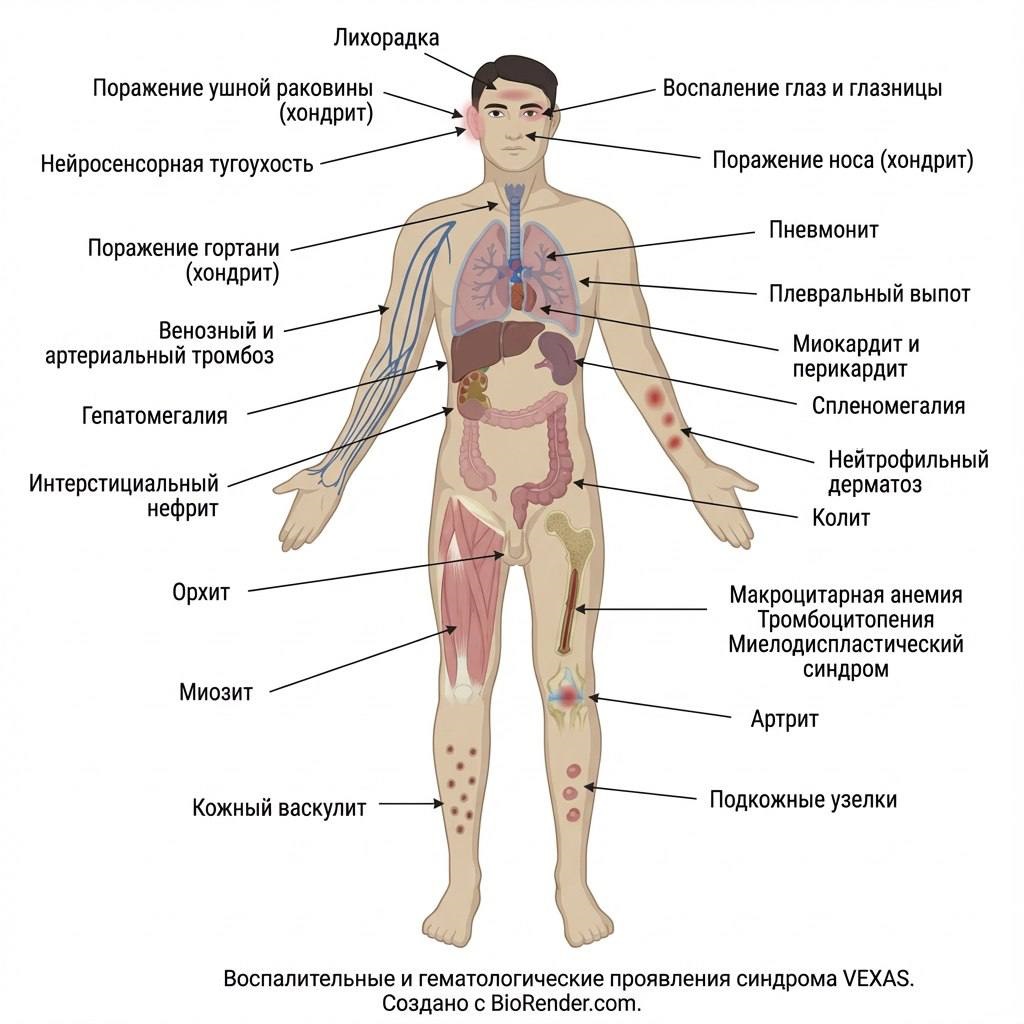
Клинические проявления заключаются в поражении кожи (нейтрофильный дерматоз, васкулит), хрящей, сосудов, легких, сердца, а также повышении температуры тела, развитии тромбозов, макроцитарной анемии, тромбоцитопении.
Лечение синдрома VEXAS сложное и требует комплексного подхода – от биологической терапии до трансплантации гемопоэтических стволовых клеток.
 Козлова Анна Леонидовна
Козлова Анна Леонидовна